ENTENDENDO A IMUNODEFICIÊNCIA PRIMÁRIA
Órgãos Maiores
Essa rede de órgãos, tecidos, células e proteínas luta contra infecções para manter seu corpo forte e saudável. ¹
Muitos invasores estranhos (“germes” ou patógenos) podem causar infecções e deixá-lo doente. ¹
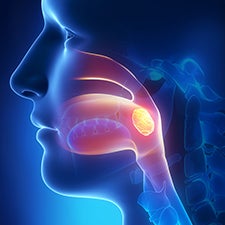
Amígdalas ¹
As amígdalas são uma dupla de órgãos linfáticos e são parte da primeira linha de defesa do sistema imune contra infecções.
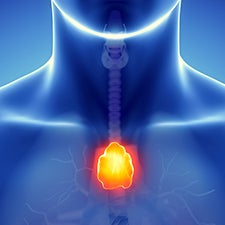
Timo ¹
Este órgão controla o desenvolvimento da maturidade de linfócitos T (células do sistema imune). Os linfócitos T são essenciais para proteção contra infecções.
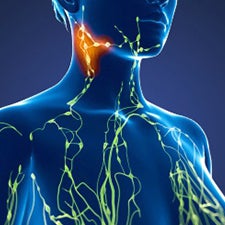
Linfonodos ¹
Você os encontrará em diversos lugares, incluindo seu pescoço e suas axilas. Os linfonodos trabalham duro para filtrar os germes que podem deixa-lo doente.
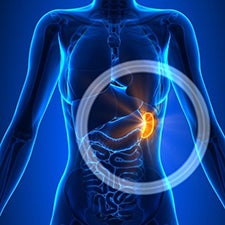
Baço ¹
O maior órgão do sistema linfático, o baço, contém leucócitos que combatem infecções e doenças. O baço também destrói leucócitos velhos e danificados enquanto ajuda a controlar a quantidade de sangue no seu corpo.
Fígado ¹
O fígado é uma grande ajuda na função imune. Conforme o sangue passa pelo fígado, suas muitas células fagocíticas (células imunes) ingerem as bactérias, células mortas ou morrendo e partículas estranhas perigosas no sangue.
Medula Óssea ¹
Uma medula óssea saudável produz células sanguíneas continuamente – eritrócitos e leucócitos e plaquetas. Esses diferentes tipos de células se desenvolvem a partir das células-tronco dentro da medula e se realocam na corrente sanguínea conforme amadurecem.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Sangue
O sangue é o componente do sistema circulatório que carrega células especializadas e proteínas do sistema imune de uma parte do corpo para outra. ¹ Algumas dessas células especializadas incluem leucócitos que são produzidos na medula óssea ou timo, como linfócitos (células B e células T) e fagócitos (neutrófilos, monócitos e macrófagos). O sistema complemento é um grupo de proteínas encontradas no sangue que são essenciais na proteção contra infecções. Quando o sistema imune não conta com essas partes, o indivíduo fica vulnerável a tipos específicos de doenças. ¹
Células T
As células T são responsáveis pela proteção de longa duração contra infecções. Há quatro tipos de células T, cada uma com um propósito especial:
- As células T auxiliares detectam infecções e deixam as outras células do sistema imune prontas para o combate. As células T auxiliares dizem às células B que elas devem produzir anticorpos, proteínas altamente especializadas que ajudam a defender o corpo contra infecções. ¹
- Células T citotóxicas/assassinas destroem as células infectadas. ¹
- As células T regulatórias dizem ao sistema imune quando a guerra com os patógenos acaba e para que parem de lutar. ¹
- As células T de memória lembram como derrotar uma infecção e podem responder rapidamente se a mesma infecção ocorrer novamente. ¹
Células B
Quando as células B encontram substâncias causadoras de doenças, elas respondem pela
maturação em células plasmáticas que produzem anticorpos. Os anticorpos são proteínas
altamente especializadas no sangue, também conhecidos como imunoglobulinas. Os anticorpos se
anexam aos invasores estranhos e os marcam para destruição. ¹
Há cinco tipos
principais de anticorpos:
- Anticorpos IgM são os primeiros a responder. Eles oferecem uma proteção importante durante os primeiros dias da infecção. Esses anticorpos tendem a ficar na corrente sanguínea, onde ajudam a eliminar as bactérias. ¹
- Anticorpos IgG são os próximos a responder. Esses anticorpos são formados em grandes quantidades e trabalham no sangue e nos tecidos do corpo. Eles se ligam aos patógenos, para que as células imunes tenham mais facilidade ao destruí-los. Os anticorpos IgG podem ser transmitidos da mãe para o bebê através da placenta. ¹
- Os anticorpos IgA são secretados nos fluidos corporais, como lágrimas, saliva e mucosas. Eles protegem contra infecções no trato respiratório e nos intestinos. Esses anticorpos podem ser transmitidos das mães para os recém-nascidos através da amamentação. ¹
- Os anticorpos IgE normalmente estão presentes em pequenas quantias e são importantes durante as reações alérgicas. ¹
- Os anticorpos IgD podem estar presentes na superfície das células B, mas sua função não é totalmente compreendida nesse momento. ¹
Fagócitos
Fagócitos são células que ingerem o patógeno ao cercá-lo e digeri-lo. Os principais tipos de fagócitos são os neutrófilos, monócitos e macrófagos. Os macrófagos atuam como sequestradores. Eles observam os patógenos invasores que possuem anticorpos ligados a eles e, então, enviam sinais de alerta para que outros macrófagos cheguem e os destruam. ¹
Sistema complemento
O sistema complemento é um grupo de proteínas na corrente sanguínea que trabalha de forma organizada para a defesa contra patógenos. Essas proteínas trabalham com anticorpos e fagócitos para livrar o corpo da infecção. ¹
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
As síndromes de imunodeficiência primária (IDP) são caracterizadas de muitas formas diferentes, incluindo:¹
- Baixos níveis de anticorpos
- Defeitos nos anticorpos
- Defeitos nas células e proteínas do sistema imune (por exemplo, células B, células T, neutrófilos ou sistema complemento)
Esses defeitos deixam as pessoas susceptíveis a infecções recorrentes e outras complicações que podem exigir terapias diferentes. Os tipos mais comuns de imunodeficiência primária resultam em uma incapacidade de produzir um tipo muito importante de proteína, chamado de anticorpos ou imunoglobulinas, que ajudam o corpo a lutar contra infecções de bactérias ou vírus. Além do aumento da susceptibilidade á infecções, as pessoas com imunodeficiência primária também podem ter doenças autoimunes, nas quais o sistema imune ataca suas próprias células ou tecidos. ¹
Imunodeficiência Comum Variável (IDCV)
Pacientes com IDCV possuem baixos níveis de imunoglobulinas séricas (anticorpos)
(hipogamaglobulinemia) ou níveis ausentes (agamaglobulinemia) e, portanto, é mais provável
que contraiam infecções. Normalmente, esses indivíduos apresentam números normais das
células que produzem anticorpos (linfócitos B), mas essas células falham na passagem da
maturação normal para as células capazes de produzir diferentes tipos de imunoglobulinas.
¹
O nome IDCV revela informações sobre a condição:
- IDCV é um tipo relativamente comum de IDP em cerca de 1 a cada 25.000 pessoas. ¹
- Muitos aspectos da IDCV são variáveis e diferem de pessoa para pessoa, incluindo o grau e o tipo de deficiência do anticorpo, a gravidade dos sinais e sintomas e o momento da vida (infância, adolescência ou fase adulta) durante o qual os sintomas surgem. ¹
- Como a IDCV pode ser diagnosticada mais tarde que outros tipos de imunodeficiência primária, alguns outros nomes que foram utilizados para ela são agamaglobulinemia adquirida, agamaglobulinemia de manifestação em adultos ou hipogamaglobulinemia de manifestação tardia. ¹
- Embora a causa exata deste distúrbio seja desconhecida, estudos recentes demonstraram que um pequeno grupo de genes parece estar envolvido para algumas pessoas com o distúrbio. ¹
Sintomas
Algumas pessoas com IDCV apresentam sintomas na infância, enquanto muitas outras podem não desenvolver os sintomas até seus 20 ou 30 anos - ou até mesmo mais tarde. Os sintomas mais comuns são infecções recorrentes envolvendo os ouvidos, sinos, nariz, brônquios (tubos respiratórios) e os pulmões. Infecções graves e repetidas dos pulmões podem resultar em alargamento permanente e cicatrizes nos brônquios - uma condição crônica chamada de bronquiectasia. ¹
Pessoas com IDCV também podem desenvolver linfonodos aumentados no pescoço, peito ou abdômen. O baço, um órgão relacionado ao sistema imune, também pode ficar maior.
Um inchaço doloroso das articulações, uma condição chamada de poliartrite, também pode se desenvolver em pessoas com IDCV. Articulações maiores como os joelhos, tornozelos, cotovelos e pulsos são afetadas com maior frequência. ¹
Queixas gastrintestinais, como dor abdominal, inchaço, náusea, vômito, diarreia e perda de peso podem ocorrer em pessoas com IDCV. ¹
Por fim, pessoas com IDCV podem apresentar maior risco de câncer, especialmente cânceres do sistema linfoide e do trato gastrintestinal. ¹
Diagnóstico e Tratamento
Os médicos suspeitam de IDCV quando uma criança ou adulto apresenta um histórico de infecções recorrentes dos ouvidos, seios da face, brônquios e pulmões. O diagnóstico é feito quando um exame sanguíneo apresenta baixos níveis de imunoglobulinas séricas, incluindo IgG, IgA e, algumas vezes, IgM. Exames laboratoriais especiais podem dizer se os linfócitos B e os linfócitos T estão trabalhando de forma adequada. Outra parte do diagnóstico de IDCV é realizada pela medição dos níveis séricos de anticorpos contra vacinas, como tétano, difteria e polissacarídeo pneumocócico. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Agamaglobulinemia ligada ao X (XLA)
XLA é uma forma hereditária de imunodeficiência primária, na qual as pessoas possuem uma mutação no gene necessário para o desenvolvimento normal de linfócitos B. O material genético responsável pela mutação está localizado no cromossomo X, por isso, o nome. ¹
Essa mutação previne que os linfócitos B se desenvolvam em células plasmáticas. Sem essas células plasmáticas, as pessoas com XLA apresentam uma deficiência grave na produção de anticorpos (imunoglobulinas). ¹
O desenvolvimento normal das células plasmáticas é um processo em etapas que começa com as células chamadas de células-tronco, que são encontradas na medula óssea. ¹
As células-tronco produzem linfócitos imaturos (leucócitos), chamados de linfócitos pró-B, que se desenvolvem para linfócitos pré-B. Esses linfócitos pré-B formam os linfócitos B ou as células B. Os linfócitos B amadurecem para células plasmáticas, que produzem imunoglobulinas específicas quando entram em contato com diferentes substâncias causadoras de doenças. ¹
Um dos primeiros tipos de IDP a ser identificado, a XLA é, algumas vezes, chamada de agamaglobulinemia de Bruton, em homenagem ao homem que a descobriu ou agamaglobulinemia congênita, em razão da incapacidade do paciente de produzir anticorpos. ¹
A XLA é transmitida em uma família como um traço recessivo ligado ao cromossomo X. Como a XLA é um distúrbio ligado ao cromossomo X, apenas garotos são afetados; no entanto, sabe-se há anos que já houve garotas com imunodeficiência que se parecia muito com XLA. Como os genes que causam a XLA foram identificados, parentes mulheres de pessoas com XLA podem ser testadas para ver se carregam o gene responsável. Em alguns casos, é possível ver se o feto de uma mulher que carrega o gene da XLA nascerá com essa condição. ¹
Sintomas
Pessoas com XLA frequentemente desenvolvem infecções que acometem os seios da face, ouvidos, brônquios ou os próprios órgãos internos. Infecções gastrintestinais, especialmente com o parasita Giardia, também podem ser um problema. Por vezes, pessoas com XLA têm problemas com infecções de pele. ¹
O exame físico da maioria dos pacientes com XLA revela amígdalas e linfonodos muito pequenos (glândulas do pescoço). Isso ocorre porque uma grande parte das amígdalas e dos linfonodos é composta por linfócitos B. Em pessoas com XLA, a ausência de linfócitos B resulta no tamanho diminuído desses órgãos. ¹
Diagnóstico e Tratamento
O diagnóstico da XLA começa com um exame de sangue que busca a presença de imunoglobulinas, pois todas as imunoglobulinas (IgG, IgM e IgA) estão ausentes ou existem apenas em pequenas quantidades na maioria das pessoas com XLA. ¹
Quando um médico suspeita que uma pessoa possui XLA, um exame de sangue adicional testará quanto ao percentual de células B, as células imunes não produzidas em XLA. Uma baixa contagem de células B no sangue é o indicador mais confiável de XLA. ¹
O diagnóstico pode ser confirmado por um teste que estabelece a ausência de uma proteína chamada de tirosina quinase de Bruton (BTK), que está associada à XLA ou pela detecção de uma anormalidade no gene BTK. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Deficiência de subclasse de IgG
Há cinco classes de imunoglobulinas (Igs): IgG, IgA, IgM, IgD e IgE. As imunoglobulinas são o mesmo que anticorpos, cuja principal função é o combate a infecções. A maioria dos anticorpos no sangue e outros fluidos são da classe IgG. A IgG é dividida em quatro subclasses: IgG1, IgG2, IgG3 e IgG4. É dito que as pessoas têm uma deficiência de subclasse de IgG quando elas não têm ou têm níveis muitos baixos de uma ou duas subclasses de IgG, mas apresentam níveis normais de outras imunoglobulinas. ¹
Anticorpos em cada subclasse reagem de forma diferente e desempenham um papel levemente diferente na proteção do corpo contra infecções. Isso significa que a ausência de uma subclasse de IgG específica deixa uma pessoa propensa a contrair certos tipos de infecções, mas não outras. Por exemplo, as subclasses de IgG1 e IgG3 são altas em anticorpos contra as toxinas produzidas por algumas bactérias, como as toxinas de difteria e tétano, bem como anticorpos contra vírus. Em contraste, anticorpos IgG2 agem predominantemente contra Streptococcus pneumoniae e Haemophilus influenzae. ¹
A IgG circulando na corrente sanguínea é composta de 60-70% IgG1, 20-30% IgG2, 5-8% IgG3 e 1-3% IgG4. Com a idade, a quantia de diferentes subclasses de IgG circulante mudará. Especialmente em crianças, esse fator deve ser considerado ao determinar se um nível de subclasse está anormal, seja em termos de ter um nível baixo da subclasse de IgG ou a incapacidade de produzir um anticorpo específico. ¹
Sintomas
Recorrência de infecções do ouvido, bronquite e pneumonia, que são as doenças mais frequentes em pessoas com deficiências de subclasse de IgG, o que podem afetar tanto homens quanto mulheres. Algumas pessoas apresentam maior frequência de infecções em idade precoce; outras só apresentarão infecções só mais tarde. Frequentemente, uma criança com deficiência de subclasse de IgG chamará a atenção do médico em razão das frequentes infecções de ouvido. Conforme a criança envelhece, podem surgir sinusite crônica ou recorrente, bronquite e pneumonia. ¹
A deficiência de subclasse de IgG2 é a deficiência de subclasse mais comum, tanto em crianças como adultos, enquanto a deficiência de subclasse de IgG3 é a deficiência mais comum observada em adultos. A deficiência de IgG4 ocorre com frequência combinada com a deficiência de IgG2. ¹
Diagnóstico e tratamento
É considerado que uma pessoa tem uma deficiência de subclasse de IgG seletiva caso os níveis sanguíneos de uma ou mais subclasses de IgG estejam abaixo da faixa normal, com base na idade, e os níveis de outras imunoglobulinas (IgG, IgA e IgM totais) estejam normais ou próximos do normal. ¹
Como uma pessoa pode ter níveis muito baixos ou nenhum de uma ou mais subclasses de IgG enquanto mantém um nível normal de IgG total, o diagnóstico requer a medição das subclasses de IgG além do IgG, IgA e IgM séricos. ¹
É importante lembrar que a definição de concentrações de subclasse de IgG “normais” variam com a idade e de laboratório para laboratório. Normalmente, os valores normais representam uma pequena faixa abaixo e acima da média para a idade de uma pessoa. ¹
Um subconjunto adicional de pacientes apresenta níveis normais de imunoglobulina e subclasses de IgG normais, ainda assim, não conseguem desenvolver níveis de anticorpos de proteção em resposta às infecções com Streptococcus pneumoniae ou vacinas contra as bactérias. Acredita-se que essas pessoas tenham uma deficiência de anticorpo específica e, normalmente, são agrupadas com pacientes com deficiência de subclasse de IgG. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Síndrome de Hiper-IgM (SHIGM)
Como o nome sugere, pessoas com a síndrome de hiper-IgM (SHIGM) apresentam um excesso de IgM
e níveis reduzidos de outras imunoglobulinas, incluindo IgG e IgA. Isso ocorre porque o
corpo não pode transformar a produção de IgM em outros anticorpos. Embora os linfócitos B do
corpo, ou células B, possam produzir IgM sozinhos, eles precisam da assistência dos
linfócitos T (ou células T), para trocar a produção de IgM para IgG, IgA ou IgE.
¹
Há uma série de defeitos genéticos que resultam na síndrome de HIGM:
- A forma mais comum da síndrome de HIGM resulta de um defeito ou deficiência de uma proteína na superfície das células T que as deixam incapazes de instruir as células B a mudar da criação de IgM para criação de outras imunoglobulinas. Chamada de síndrome de HIGM ligada ao X (XHIGM), essa forma da síndrome de HIGM é transmitida como um traço recessivo ligado ao X e normalmente é encontrada apenas em garotos. ¹
- Outras formas da síndrome de HIGM são herdadas tanto por garotos quanto por garotas como traços recessivos autossômicos. Como as formas recessivas autossômicas de HIGM precisam que o gene em ambos os cromossomos seja afetado, elas são menos frequentes que as condições ligadas ao X.¹
- O último tipo de síndrome de HIGM está associado com uma condição de pele chamada de displasia ectodérmica, na qual os pacientes apresentam cabelos ralos e dentes coniformes e infecções recorrentes. Este é outro traço ligado ao X que afeta uma molécula necessária para comutação do sinal de produção de imunoglobulina. ¹
Sintomas
A maioria das crianças com síndrome de HIGM desenvolverá os sintomas entre o 1º e o 2º ano de vida. O problema mais comum é um aumento no risco de infecções, incluindo infecções repetidas do trato respiratório superior e inferior. A maioria das infecções é bacteriana, mas também podem ser virais ou fúngicas. Alguns pacientes também desenvolvem problemas gastrintestinais, incluindo diarreia ou má absorção. ¹
Garotos com síndrome de XHIGM podem desenvolver uma forma oportunista de pneumonia ou uma infecção gastrintestinal que pode provocar uma doença grave no fígado. Cerca de metade dos pacientes com XHIGM desenvolve uma neutropenia com baixa contagem de leucócitos, que está associada a úlceras da boca, inflamações ou ulceração do reto e infecções de pele. ¹
Pacientes com HIGM recessivo autossômico são mais propensos a apresentarem crescimento dos linfonodos do que pacientes com outros tipos de imunossupressão primária. Como resultado, frequentemente, essas pessoas apresentam aumento das amígdalas e adenoides que podem causar ronco e apneia obstrutiva do sono. Doenças autoimunes, quando o sistema imune de uma pessoa ataca o corpo, também podem ocorrer em pacientes com HIGM. ¹
Diagnóstico e tratamento
A síndrome de HIGM é suspeita em um paciente apresentando infecções respiratórias graves recorrentes ou uma infecção oportunista quando exames de sangue demonstrarem um nível normal ou elevado de IgM e baixo ou zerado de IgG. O diagnóstico final é baseado em uma análise do DNA da pessoa para identificar mutações nos genes conhecidas por causar a síndrome de HIGM. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Deficiência Seletiva de IgA
Indivíduos com deficiência seletiva de IgA não possuem IgA, mas normalmente apresentam quantias normais de outras imunoglobulinas. A maioria das pessoas afetadas não fica doente como resultado da deficiência; outras podem desenvolver uma diversidade de problemas graves. No entanto, muitos não são diagnosticados, pois nunca ficam doentes o suficiente para procurar um médico. ¹
A IgA protege o corpo nas superfícies que entram em contato com o meio ambiente. Esses locais são as superfícies mucosas - boca, ouvidos, sinos, nariz, garganta, passagens de ar dentro dos pulmões, trato gastrintestinal, olhos e genitais. ¹
Anticorpos IgA são transportados em secreções para essas superfícies mucosas e desempenham um papel na proteção contra infecções, e é por isso que a IgA é conhecida como um anticorpo secretório. Como a área das superfícies mucosas de uma pessoa é igual a 1,5 quadra de tênis, a importância de IgA na proteção dessas superfícies não pode ser superestimada. ¹
Embora as pessoas com deficiência seletiva de IgA não produzam IgA, elas produzem todas as outras imunoglobulinas. Além disso, os outros aspectos de seus sistemas imunes funcionam de forma adequada. As causas da deficiência seletiva de IgA permanecem desconhecidas. É provável que seja uma série de causas que variam de pessoa para pessoa. ¹
IgA sérico baixo, como IgA sérico ausente, é relativamente comum. A maioria das pessoas com IgA sérico baixo não tem doença aparente; outras apresentam sintomas semelhantes à imunodeficiência comum variável (IDCV). ¹
Sintomas
A deficiência seletiva de IgA é um dos tipos mais comuns de imunodeficiência primária (estima-se 1 em cada 500 caucasianos). Muitas pessoas com deficiência seletiva de IgA parecem saudáveis ou apresentam doenças relativamente leves, o que significa que elas podem nunca ser diagnosticadas. Ao mesmo tempo, há pessoas com deficiência seletiva de IgA que estão muito doentes. ¹
Pessoas com deficiência seletiva de IgA correm o risco de infecções. Cerca de metade dos pacientes com deficiência seletiva de IgA que chamam a atenção do médico apresentam repetidas infecções nos ouvidos, sinos e brônquios (passagens de ar dos pulmões) e pneumonia. Alguns outros pacientes com deficiência seletiva de IgA apresentam infecções gastrintestinais e diarreia contínua. Essas infecções comuns afetam as áreas com superfícies mucosas que a IgA protegeria e podem se tornar crônicas. Algumas vezes, as infecções não desaparecem totalmente com o tratamento e os pacientes podem precisar de antibióticos por mais tempo que o normal. ¹
Um segundo problema importante na deficiência seletiva de IgA é a ocorrência de doenças autoimunes. Elas são encontradas em cerca de 25% a 33% dos pacientes com deficiência seletiva de IgA que visitam o médico. Em doenças autoimunes, as pessoas produzem anticorpos que atacam seus próprios tecidos e resultam em inflamações e lesões. ¹
As alergias podem ser mais comuns entre pessoas com deficiência seletiva de IgA do que na população geral, ocorrendo em cerca de 10 a 15% desses indivíduos. Os tipos de alergia variam. A asma é uma das doenças alérgicas mais comuns que ocorrem com a deficiência seletiva de IgA. Alergia a alimentos também pode estar associada à deficiência seletiva de IgA. ¹
Comumente, pessoas com deficiência seletiva de IgA são consideradas com maior risco de reações anafiláticas ou com risco de vida quando recebem derivados do sangue, incluindo tratamento IVIg que contém alguma IgA. ¹
Diagnóstico e tratamento
Infecções contínuas ou frequentes, alergias, doenças autoimunes e diarreia crônica, ou uma combinação desses problemas, podem levar os médicos a suspeitarem da deficiência seletiva de IgA. O diagnóstico é feito quando os exames laboratoriais mostram que o sangue de uma pessoa apresenta uma ausência de IgA com níveis normais dos outros principais tipos de imunoglobulinas (IgG e IgM). ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Imunodeficiência Combinada Grave (SCID)
Pronunciado “skid”, SCID é uma síndrome rara com múltiplas causas genéticas, e normalmente é considerada a forma mais séria da imunodeficiência primária. Atualmente, há 12 causas genéticas conhecidas de SCID. Essas causas genéticas diferem em relação à mutação exata que causa a SCID e aos padrões de hereditariedade, mas em comum apresentam deficiências graves na função das células T e das células B. Para muitas pessoas, outras células no sistema imune também estão envolvidas. ¹
Sintomas
Uma alta taxa de infecções é o sintoma mais comum em bebês com SCID. Essas infecções não são os mesmos tipos de infecções, como resfriados, apresentados por crianças com sistemas imunes normais. Crianças com SCID desenvolvem infecções graves e mesmo com risco de morte, e podem incluir pneumonia, meningite ou infecções na corrente sanguínea. O uso difundido de antibióticos deixou ainda mais difícil para os médicos identificarem a SCID. ¹
Crianças com SCID estão vulneráveis às mesmas infecções que afetam bebês com sistemas imunes saudáveis. Além disso, elas correm risco de desenvolver infecções causadas por organismos, como o vírus da catapora (varicela) ou vacinas que não são prejudiciais em crianças saudáveis. Um dos mais perigosos é o Pneumocystis jirovecii, o que pode causar uma forma rapidamente fatal de pneumonia em pacientes com SCID se não diagnosticados e tratados rapidamente. ¹
Crianças com SCID também estão vulneráveis a infecções fúngicas difíceis de tratar, como candidíase oral, uma infecção por Cândida (fungos) na boca. Essas infecções podem melhorar, mas não irem embora, ou podem voltar assim que a medicação parar. ¹
Além disso, crianças com SCID podem desenvolver diarreia persistente, que pode levar a falha no crescimento e má nutrição. ¹
No Brasil, esses pacientes podem desenvolver reações graves à vacinação contra tuberculose (BCG), realizada no primeiro mês de vida. Essas reações incluem disseminação da doença e reações locais severas à vacina. ²
Diagnóstico e tratamento
Normalmente, SCID tem sua primeira suspeita em crianças quando elas desenvolvem infecções graves associadas com SCID. Em alguns casos, o diagnóstico é suspeito por haver outra criança na família com SCID. ¹
Dois exames de sangue são utilizados para se chegar a um diagnóstico. O primeiro é um hemograma completo. O segundo teste permite que o médico calcule o número de linfócitos no sangue. Caso a contagem ainda seja baixa, testes adicionais são conduzidos para contagem das células T e medição de função das células T para confirmar o diagnóstico. ¹
Em alguns casos, SCID pode ser diagnosticada antes mesmo do bebê nascer. Caso uma análise tenha determinado qual mutação é responsável por uma SCID de irmãos, o teste genético pode ser realizado em células coletadas a partir de amostras das vilosidades coriônicas (CVS) ou da amniocentese. ¹
O diagnóstico precoce de SCID, antes do bebê ter uma chance de desenvolver qualquer infecção, é muito importante. Caso um transplante de medula óssea seja realizado durante os primeiros três meses de vida, a taxa de sucesso é de 94%. Com o rastreamento de rotina, quase todos os bebês com SCID poderiam ser diagnosticados nos primeiros dias de vida. ¹
Vivendo com SCID
Bebês com SCID precisam ficar isolados de crianças fora da família. Caso haja irmãos que frequentem uma creche, escola ou realizem outras atividades, a possibilidade de trazer a catapora para casa é o maior perigo. Isso tem diminuído devido ao uso difundido da vacina contra catapora, porém os pais ainda devem avisar à escola que eles precisam ser notificados caso o vírus da catapora esteja presente na escola. ¹
Crianças que receberam a vacina contra pólio com o vírus vivo podem propagar o vírus para uma criança com SCID, então é aconselhável que irmãos recebam a vacina contra pólio com vírus inativo. ¹
Crianças com SCID não devem receber vacinas vivas, incluindo rotavírus, catapora, caxumba, sarampo, pólio com vírus vivo ou vacinações contra BCG até que sua situação imune seja avaliada. Os irmãos não devem receber a vacina viva do rotavírus. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referências: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013. 2. Mazzucchelli, J. T. L. et al. Severe Combined Immunodeficiency in Brazil: Management, Prognosis, and BCG-Associated Complications. J Investig Allergol Clin Immunol 24, 184-191 (2014).
Síndrome de Wiskott-Aldrich (WAS)
A síndrome de Wiskott-Aldrich é uma imunodeficiência primária que afeta tanto os linfócitos T
quanto B. Além disso, as plaquetas (células sanguíneas que ajudam a controlar sangramentos)
estão reduzidas em número e tamanho. Pessoas com WAS apresentam frequentes infecções
bacterianas, virais e fúngicas; eczema da pele e uma maior tendência a sangramentos. WAS é
causada por mutações ou erros no gene que produz uma proteína com o nome do distúrbio, a
proteína da síndrome de Wiskott-Aldrich (WASP), que está localizada no cromossomo X. Como o
defeito do gene é carregado no cromossomo X, apenas garotos podem herdar WAS.
¹
Pessoas com WAS demonstraram um padrão clássico nos sintomas:
¹
- Maior tendência a sangramentos, causados por um número significativamente reduzido das plaquetas.
- Infecções bacterianas, virais e fúngicas recorrentes.
- Eczema da pele.
Sintomas
Indivíduos com WAS podem desenvolver diferentes sintomas. Algumas pessoas apresentam os três sintomas clássicos; outras apresentam baixas contagens de plaquetas e sangramento. Todos estão relacionados com a mutação no gene WAS. ¹
Sangramento:
Uma redução no tamanho e no número de plaquetas é uma marca da WAS. Essas plaquetas pequenas são exclusivas de WAS e sua presença é útil na realização do diagnóstico. O sangramento na pele pode provocar pontos vermelhos-azulados (chamados de petéquias) variando no tamanho, de pequenos alfinetes a grandes hematomas. Garotos com WAS também podem apresentar movimentos intestinais com sangue (especialmente na infância), sangramento na gengiva e sangramentos nasais prolongados. Hemorragias no cérebro são complicações especialmente perigosas. ¹
Infecções:
Devido à deficiência da função dos linfócitos B e T, as infecções são comuns em WAS. Normalmente, essas infecções envolvem os seios da face, ouvidos e pulmões, incluindo pneumonia. Infecções mais graves da corrente sanguínea também podem ocorrer, bem como meningite ou infecções virais graves. ¹
Eczema:
Pessoas com WAS clássico frequentemente sofrem com eczema. Em bebês, o eczema pode parecer uma “crosta láctea” ou irritação da fralda grave; em garotos mais velhos, ela pode estar limitada às marcas na pele ao redor da frente do cotovelo, ao redor do pulso e do pescoço e atrás do joelho. ¹
Manifestações autoimunes:
Uma alta incidência de sintomas “do tipo autoimune” (condições que resultariam do sistema imune não saudável atacando o próprio corpo do paciente) é comum tanto em bebês quanto em adultos com WAS. O mais comum desses é a vasculite, uma inflamação dos vasos sanguíneos; anemia hemolítica, quando o corpo destrói suas próprias células sanguíneas e púrpura trombocitopênica idiopática (PTI), quando o sistema imune ataca as plaquetas. ¹
Câncer:
Câncer pode ocorrer em garotos e homens com WAS. A maioria desses cânceres, como linfoma ou leucemia, envolve as células B.¹
Diagnóstico e tratamento
Devido às anormalidades das plaquetas de WAS estarem quase sempre presentes, mesmo no cordão umbilical no nascimento, a forma mais fácil de diagnosticar WAS é obter uma contagem de plaquetas e determinar o tamanho das plaquetas. As plaquetas de WAS são significativamente menores que as plaquetas normais. Em crianças com mais de dois anos, a frequência das infecções e deficiências no sistema imune pode ser utilizada para sustentar o diagnóstico. ¹
O diagnóstico é confirmado ao demonstrar uma ausência da proteína WAS nas células sanguíneas ou pela identificação de uma mutação dentro do gene WAS. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Ataxia Telangiectasia (A-T)
Ataxia telangiectasia (A-T) é uma doença da imunodeficiência primária que afeta diversos órgãos. A-T pode ser herdada e o gene responsável ser identificado. Entre os sintomas, está um sistema imune enfraquecido, que envolve tanto os linfócitos T quanto B, resultando no aumento da susceptibilidade a infecções. ¹
Ela é caracterizada por anormalidades neurológicas que resultam em um caminhar instável (ataxia), vasos sanguíneos dilatados (telangiectasia) nos olhos e na pele, defeitos imunes envolvendo linfócitos T e B e uma predisposição ao câncer. ¹
Sintomas
Normalmente, o primeiro sintoma é ataxia, um termo médico utilizado para descrever uma marcha instável, que aparece quando uma criança começa a andar. Outros sintomas relacionados às anormalidades do cérebro incluem problemas com os movimentos dos olhos, como piscadas alternadas rapidamente dos olhos e dificuldade no controle do movimento dos olhos; e dificuldade de utilizar os músculos necessários para fala e deglutição. ¹
Vasos sanguíneos dilatados fazem com que a parte branca dos olhos apareça vermelha. Também podem ser observados com menos frequência nos ouvidos, pescoço e nas extremidades. ¹
Um risco maior de infecções causadas por bactérias e vírus também é observado em pessoas com A-T, afetando principalmente os pulmões e os seios da face. Parte desse aumento no risco de infecções está relacionada à dificuldade de deglutição (disfagia), que pode levar à aspiração com alimentos e líquidos indo para os pulmões, e não para o estômago. ¹
Diagnóstico e tratamento
O diagnóstico de A-T é normalmente feito com base nas características clínicas comuns
(ataxia, telangiectasia, movimento anormal dos olhos e fala). Testes laboratoriais podem
ajudar a chegar ao diagnóstico, especialmente na primeira infância, quando a telangiectasia
não apareceu. ¹
Os testes laboratoriais incluem:¹
- Medição da alfafetoproteína, pois níveis elevados são encontrados na maioria dos pacientes com A-T após os 18-24 meses de idade.
- Ausência da proteína produzida pelo gene A-T também pode confirmar o diagnóstico.
- Medição dos danos às células após exposição no raio-x.
- Exame do gene A-T para buscar erros genéticos (mutações).
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Síndrome de DiGeorge
A síndrome de DiGeorge é uma forma de IDP causada pela formação anormal de alguns tecidos durante o desenvolvimento fetal. O defeito pode afetar a glândula do timo e prejudicar a produção dos linfócitos T.¹
A maioria das pessoas com síndrome de DiGeorge apresenta um defeito genético no cromossomo 22. Isso leva à anormalidade nas células e no desenvolvimento do tecido, resultando em defeitos nos órgãos afetados. O envolvimento do órgão e a gravidade do envolvimento do órgão podem variar entre os indivíduos. ¹
Sintomas
Os indivíduos com síndrome de DiGeorge podem apresentar um ou todos os sintomas a seguir:
-
Aparência facial:
Crianças podem ter um queixo subdesenvolvido, olhos com pálpebras pesadas, orelhas viradas para trás e partes superiores com defeito em seus lóbulos auriculares. ¹
-
Anormalidades da glândula paratireoide:
Uma glândula paratireoide subdesenvolvida pode provocar o hipoparatireoidismo, levando à dificuldade na manutenção dos níveis normais de cálcio, chamada de hipocalcemia. Isso pode causar tonturas ou convulsões. O defeito da paratireoide frequentemente fica menos grave com o passar do tempo. ¹
-
Defeitos do coração:
A maioria dos defeitos cardíacos afeta a aorta (vaso sanguíneo que retira o sangue rico em oxigênio do coração para o corpo) ou a parte do coração na qual a aorta se desenvolve. Alguns pacientes podem ter anomalias cardíacas graves, enquanto outros podem não ter nenhuma. ¹
-
Anormalidades da glândula do timo:
O subdesenvolvimento ou o desenvolvimento incompleto da glândula do timo resulta em um timo com tamanho menor que o normal e produção reduzida das células imunes, chamadas de linfócitos T ou células T. As células T não apenas destroem as células infectadas, como também ajudam os linfócitos B (células B) a se desenvolverem em células plasmáticas e a produzirem imunoglobulinas ou anticorpos. Isso resulta em um aumento do risco de infecções virais, fúngicas e bacterianas.¹
-
Autoimunidade:
Pacientes com síndrome de DiGeorge desenvolvem doenças autoimunes em uma taxa mais alta que a população geral. A doença autoimune ocorre quando o sistema imune ataca, de forma inadequada, seu próprio corpo. ¹
-
Outras características clínicas:
Indivíduos com a síndrome de DiGeorge também podem apresentar outras anormalidades de desenvolvimento, incluindo palatina, função fraca do palato, atraso na fala e dificuldade na alimentação e deglutição. Algumas pessoas também apresentam dificuldades na aprendizagem, problemas de comportamento, distúrbios psiquiátricos e hiperatividade. ¹
Diagnóstico e tratamento
Tradicionalmente, a síndrome de DiGeorge era diagnosticada quando uma pessoa apresentava as características descritas acima. No entanto, pessoas com síndrome de DiGeorge diferem não apenas nos órgãos e tecidos afetados, mas também em termos da gravidade com que um órgão ou tecido é afetado. Isso significava que muitos casos leves foram perdidos quando o diagnóstico se baseava apenas nos sintomas. ¹
Nos últimos anos, o diagnóstico foi baseado em um teste genético que pode detectar a seção excluída do cromossomo 22, que é encontrado em 90% dos indivíduos com síndrome de DiGeorge. A pequena proporção de pacientes sem o defeito genético é diagnosticada com base nas características clínicas. ¹
Apenas seu médico pode fazer seu diagnóstico e determinar qual tratamento é correto para você e suas necessidades de saúde específicas.
Referência: 1. Blaese RM, Bonilla FA, Stiehm ER, Younger ME, eds. Patient & Family Handbook for Primary Immunodeficiency Diseases. 5a ed. Towson, MD: Fundação da Imunodeficiência; 2013.
Quais são os sinais de alerta da IDP em adultos?
Esses sinais de aviso foram desenvolvidos pelo Painel Consultor da Fundação Médica Jeffrey Modell. É altamente recomendado o aconselhamento com especialistas em Imunodeficiência Primária. ¹
Caso você ou alguém que você conheça tenha dois ou mais dos sinais de alerta a seguir, converse com um médico sobre a possível presença de um distúrbio subjacente da imunodeficiência primária e um possível diagnóstico de imunodeficiência primária. ¹
- Duas ou mais novas infecções de ouvido em um ano.
- Duas ou mais novas infecções dos seios da face (sinusites) em um ano, na ausência de alergias.
- Uma pneumonia por ano por mais de um ano.
- Diarreia crônica com perda de peso.
- Infecções virais recorrentes (resfriados, herpes, verrugas, condiloma).
- Necessidade recorrente de antibióticos intravenosos para curar infecções.
- Abscessos recorrentes e profundos da pele ou dos órgãos internos.
- Candidíase persistente ou infecção fúngica na pele ou em outro lugar.
- Infecção com bactérias do tipo de tuberculose.
- História familiar de imunodeficiência primária.

Referência: 1. Painel Consultor da Jeffrey Modell Foundation. 10 Sinais de Aviso da Imunodeficiência Primária. Jeffrey Modell Foundation website. 2013. http://www.info4pi.org/library/educational-materials/10-warning-signs. Último acesso em setembro de 2016.
Quais são os sinais de alerta da IDP em crianças?
Esses sinais de aviso foram desenvolvidos pelo Painel Consultor da Fundação Médica Jeffrey Modell. É altamente recomendado o aconselhamento com especialistas em Imunodeficiência Primária. ¹
Caso uma criança tenha dois ou mais dos sinais de alerta a seguir, converse com um médico sobre a possível presença de um distúrbio subjacente da PID e um possível diagnóstico de imunodeficiência primária. ¹
- Duas ou mais novas infecções de ouvido em um ano;
- Duas ou mais novas infecções graves dos seios da face (sinusites) em um ano na ausência de alergias;
- Dois ou mais meses em antibióticos com pouco efeito;
- Duas ou mais pneumonias em um ano;
- Incapacidade de um bebê de ganhar peso ou crescer normalmente;
- Abscessos recorrentes e profundos da pele ou dos órgãos;
- Candidíase persistente na boca ou infecção fúngica na pele;
- Necessidade de antibióticos intravenosos para curar infecções;
- Duas ou mais infecções profundas, incluindo septicemia;
- História familiar de imunodeficiência primária.

Referência: 1. Painel Consultor da Jeffrey Modell Foundation. 10 Sinais de Aviso da Imunodeficiência Primária. Jeffrey Modell Foundation website. 2013. http://www.info4pi.org/library/educational-materials/10-warning-signs. Último acesso em setembro de 2016.